Last week saw the revised version of the Notified Bodies Survey on certifications and applications (MDR/IVDR) published by the European Commission. [Survey NBs availability (europa.eu)]
While there are still no conclusions made from the results of the survey, it does offer a useful insight on the current state of play for the transition to the MDR for medical devices and IVDR for in-vitro diagnostics.
The challenges the industry has faced to achieve compliance to the new regulations has been widely discussed [refer to previous blogs], resulting in continued delays to the implementation of the regulations, based largely on the state of readiness of the regulatory infrastructure of the EU system.
The regulation has been designed “to ensure the smooth functioning of the internal market as regards in vitro diagnostic medical devices, taking as a base a high level of protection of health for patients and users, and taking into account the small and medium-sized enterprises that are active in this sector.”
From <https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32017R0746&qid=1685433681978>
In reality, the challenge for SMEs to comply to the new IVDR is great, given the additional data burden, time, and money required to obtain a CE mark. I have often wondered if the EU market is a step too far as a 1st launch country for SMEs, over the opportunity of US market access – and the UK (if the regulators can identify a pragmatic approach under IDAP).
It is therefore an interest to see the graph shown in the report:
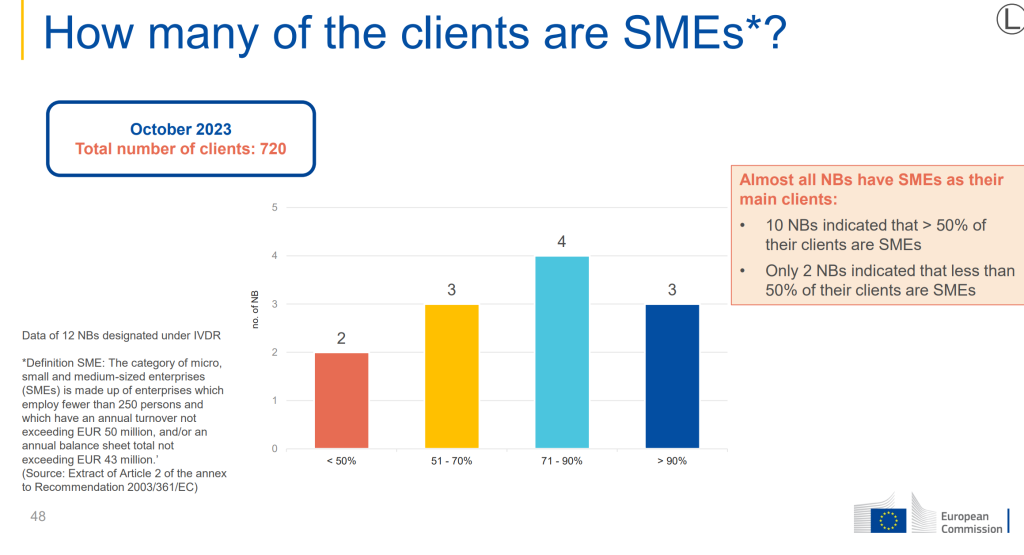
The survey highlighted that the NB have SMEs as their main clients for conformity assessment, this is a positive message in that SMEs are still preparing for IVDR compliance, and it will be interesting to see how the implementation of IVDR is achieved for small and medium size businesses.
Here at IVDeology, an immediate question that came from the information above is that the definition of SME encompasses a large number of organisations but internally the teams can vary in size, and the ones we commonly work with here at IVDeology would be at the lower end of the scale. It would be good to know the spread of size of SME’s out there so that we can understand the landscape and dynamic of those hoping to grow.
IVDeology works throughout the whole IVD industry, including supporting SMEs along their path to compliance. For further information on how we can support you, contact [email protected]