Understanding and Bridging the Gap in Clinical Research
The European Union’s (EU) healthcare landscape is continuously evolving, driven by the need to integrate
innovative treatments and technologies. One of the most ambitious initiatives in this realm is the EU
COMBINE Programme, launched in June 2023. This programme aims to address the complexities
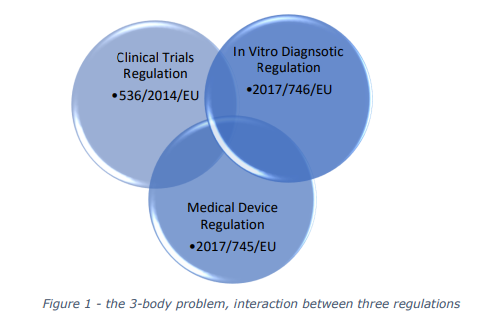
and challenges at the intersection of three critical regulations: the Clinical Trials Regulation (CTR),
the Medical Devices Regulation (MDR), and the In Vitro Diagnostic Medical Devices Regulation
(IVDR).
Understanding the Need for COMBINE
In the EU, clinical trials of medicinal products, clinical investigations of medical devices, and
performance studies of in vitro diagnostics (IVDs) are governed by distinct regulatory frameworks.
These regulations, while comprehensive, often create operational challenges when combined
studies are required. Combined studies involve:
- A clinical trial of a medicinal product alongside a performance study of an IVD
- A clinical trial of a medicinal product alongside a clinical investigation of a medical device
The COMBINE Programme was initiated to streamline these processes, ensuring that innovative
treatments combining medicinal products with medical devices or IVDs can be developed more
efficiently.
One of the challenges presented by the stakeholders, was the duplication of work by submitting
combined study application across multiple countries within the EU. Studies spanning multiple
countries require multiple applications to each competent authority. In some cases, the information
required, and outcome can vary depending on the country. Not only is this a duplication of work, it
can put clinical study progression at risk.

The Goals of the COMBINE Programme
The primary objectives of the COMBINE Programme are to:
- Analyse Challenges: Identify and understand the root causes of difficulties faced by sponsors
in conducting combined studies. - Propose Solutions: Develop practical solutions to address these challenges, facilitating
smoother regulatory processes.
Key Phases of the Programme
The COMBINE Programme is structured into two main phases:
- Analysis Phase: This initial phase involved collecting and analysing feedback from various
stakeholders, including competent authorities, medical research ethics committees, and the
European Medicines Agency (EMA). The findings were published in an analysis report in May
2024, highlighting the primary issues and proposing potential solutions. - Implementation Phase: Following the analysis, the Member States endorsed a strategy for
the second phase in December 2024. This phase focuses on implementing the proposed
solutions through a series of projects. These projects aim to align the regulatory frameworks
and simplify the processes for combined studies.
Stakeholder Involvement
The success of the COMBINE Programme hinges on the collaboration of a diverse group of
stakeholders, including:
- Competent Authorities: National bodies responsible for overseeing clinical trials and
medical devices. - Medical Research Ethics Committees: Groups ensuring that clinical studies meet ethical
standards. - European Medicines Agency (EMA): The agency providing scientific evaluation, supervision,
and safety monitoring of medicines in the EU. - Industry Representatives: Stakeholders from the pharmaceutical and medical device sectors, including associations like the European Federation of Pharmaceutical Industries and Associations (EFPIA) and MedTech Europe.
Progress and Future Directions
As of early 2025, the projects within the COMBINE Programme are progressing according to the
established timelines. Regular reviews and updates ensure that the programme remains on track
and responsive to emerging challenges.
The ultimate goal is to create a more cohesive and efficient regulatory environment that supports
the development of innovative treatments, benefiting patients across the EU
The role of EUDAMED
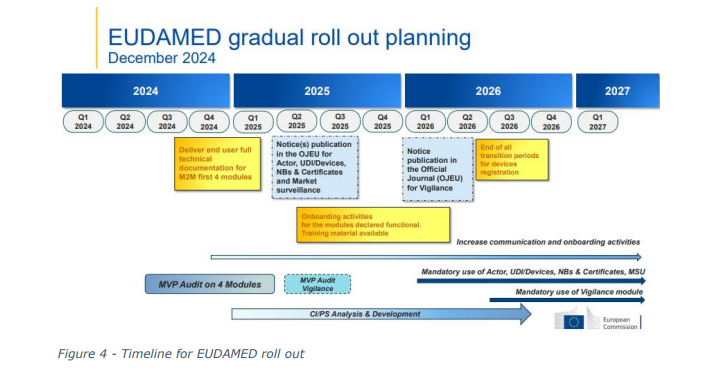
EUDAMED (European Database on Medical Devices) is an IT system developed by the European
Commission to implement the Medical Devices Regulation (MDR) and the In Vitro Diagnostic Medical
Devices Regulation (IVDR). With the transition to the new regulations, EUDAMED will be enhanced
to create a more transparent and efficient regulatory tool for all industry stakeholders. However, the
implementation of EUDAMED has seen a number of delays, largely due to the complexity of the
system and resources required to implement. The result has been a lack of coordination between
competent authorities, this can be attributed to some of the challenges identified as part of the
COMBINE programme.
Conclusion
The EU COMBINE Programme represents a significant step forward in harmonizing the regulatory
landscape for combined studies. By addressing the complexities at the intersection of CTR, MDR, and
IVDR, the programme aims to foster innovation and improve patient access to cutting-edge
treatments.
The eventual roll out of EUDAMED will enable greater transparency and communication between
member states, and regulatory authorities.
As the projects within the programme continue to unfold, the healthcare community remains
optimistic about the potential for streamlined processes and enhanced collaboration.
Stay tuned for more updates on the progress and impact of the COMBINE Programme as it continues
to shape the future of clinical research in the EU.
How IVDEOLOGY is supporting the industry
The IVD regulation is a key cog in the mechanism for combined studies. We work with a number of
manufactures developing Companion Diagnostic medical devices, or clinical trial assays which fall
under this regulation. We help build regulatory strategies, and technical documentation to support
regulatory submissions.
Book a call with us to discuss your regulatory challenges or questions and see how IVDeology can
help you
References: