Laboratory Developed Tests (LDT) have been used for decades. However, it is clear that regulatory authorities have seen the misuse of these tests. They are now moving to close the loopholes in regulation that have exploited over the years and the implementation of the European IVD Regulation (2017/746) introduced those controls.
On 29 April 2024, the US FDA published the Final Rule amending the US Food, Drug and Cosmetic Act (FD&C). There are two main purposes for this update:
• To formally identify that IVDs are devices under the FD&C Act, including if they are manufactured in a laboratory but updating the definition of an IVD.
• To phase out the general enforcement discretion approach for LDTs over 4 years. This means that they will now generally fall under the same regulation and enforcement approach as other IVDs.
This update was long overdue, curiously the last time LDTs had been considered was under the Medical Device Amendments of 1976 which amended the FD&C act to ensure the regulation of devices intended for human use. Since then, the FDA has exercised enforcement discretion for LDTs meaning that laboratories designing and manufacturing LDTs did not need to comply with those requirements.

Exceptions to the Rule
The change in rules do not apply to all devices, the following exemptions have been included for some circumstances:
LDTs marketed before Final Rule was published on 06 May 2024
- Laboratory must still have Medical Device Reporting in place.
- Must have Establishment and Device Listing in place.
- Must comply with record keeping requirements.
Tests meeting unmet needs, Nonmolecular antisera LDTs
- Record keeping requirements must be followed.
- Other aspects of QMS are expected.
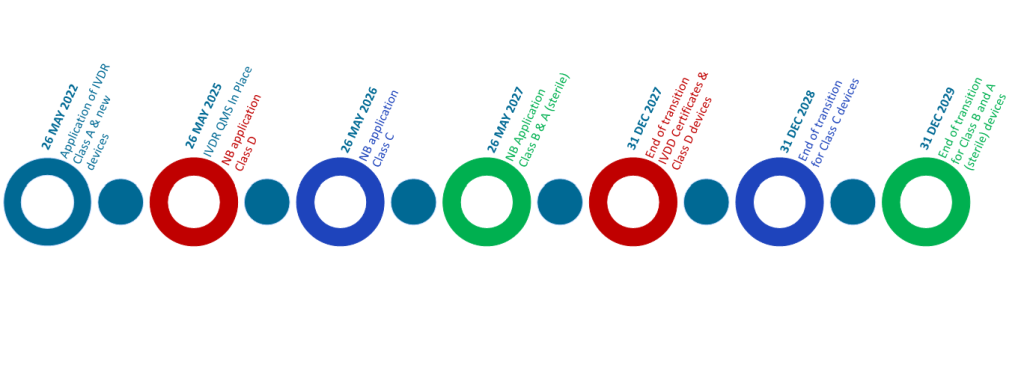
Understanding the Transitional Timelines
The transition will be a graduated process over the next three years. It is important that manufacturers understand what changes are expected, and when they are required.

Conclusion
Bringing LDTs under regulatory control can only be a good thing, the increase in high-risk, high-volume tests being offered to the public e.g. cancer risk prediction under LDT umbrella has forced regulators to act. There is no doubt that the future enforcement of regulatory controls will mean increased costs and resource requirements for those laboratories manufacturing LDTs in the US.
Unlike the EU IVD Regulation, it is clear that the FDA is using a less burdensome approach providing exceptions to the requirement to formally submit applications and wait for approvals if the LDT was already on “on market” before 06 May 2024. This does seem to be their current direction of travel in that they are actively seeking to down classify devices from PMA to 510(K) and to implement ISO 13485 to align Quality Management System requirements going forward.
Control is clearly moving to laboratories offering this type of test having QMS procedures in place to ensure that records are kept, and traceability is maintained. Oversight will be delivered with quality audits identifying issues and Medical Device Reporting highlighting issues to the FDA. The FDA will have the ability to show up on the doorstep of any facility they believe is having issues and this may be identified via market surveillance and/or patient/physician reporting of issues.
Next steps to consider and how we can help

IVDeology Ltd can support with all of the above, please contact us for a friendly conversation to identify how we can support you with your compliance journey by clicking here
By Nancy Consterdine, Co-founder and Director of Training at IVDeology Ltd and UKRP